A16A Autres produits pour les voies digestives et le métabolisme

Langue
- Français
Français
Langue
-
Français
 Médicaments en Russie
Médicaments en Russie
таб., покр. кишечнораствор. обол.:
400 мг
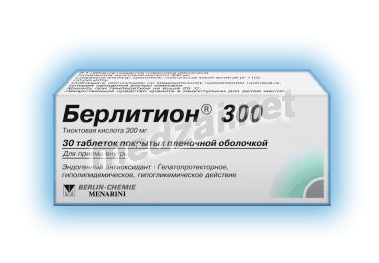
таб., покр. плен. обол.:
300 мг
концентрат д/пригот. р-ра д/инфузий:
25 мг/мл
концентрат д/пригот. р-ра д/инфузий:
1 мг/мл

таб., покр. кишечнораствор. обол.:
500 мг
лиофилизат д/пригот. р-ра д/инфузий:
400 ЕД, 200 ЕД
 Médicaments en France
Médicaments en France
comprimé dispersible:
200 mg
comprimé dispersible:
200 mg
poudre pour solution buvable et solution gastro-entérale:
10 g, 5 g

solution buvable:
1,89 g, 2 g
comprimé effervescent:
1,89 g, 1,89 mg, 1,890 g, 2 g
poudre pour solution buvable:
2 g
granulés:
10 g
granulés pour suspension buvable:
9,45 g
comprimé effervescent:
1,890 g
solution à diluer pour perfusion:
2 mg
 Русский
Русский